Typically, you’ll want to use a validation set to determine an optimal threshold as it is often not .5 (which is equivalent to argmax). Subsequently, use this threshold on the the “_prob” image to generate a binary image.This blog posts explains how to train a deep learning lymphocyte detector in accordance with our paper “Deep learning for digital pathology image analysis: A comprehensive tutorial with selected use cases”.
Please note that there has been an update to the overall tutorial pipeline, which is discussed in full here.
This text assumes that Caffe is already installed and running. For guidance on that you can reference this blog post which describes how to install it in an HPC environment (and can easily be adopted for local linux distributions).
Preface
As mentioned in the paper, the goal of this project was to create a single deep learning approach which worked well across many different digital pathology tasks. On the other hand, each tutorial is intended to be able to stand on its own, so there is a large amount of overlapping material between Use Cases.
If you’ve already read the Use Case 1: Nuclei Segmentation, in this tutorial you can focus on “Step 1: Patch Extraction”, which contains all the differences from the nuclei tutorial.
Background
Lymphocytes, a sub-type of white blood cells, are an important part of the immune system. Lymphocytic infiltration is the process by which the density of lymphocytes greatly increases at sites of disease or foreign bodies, indicating an immune response. A stronger immune response has been highly correlated to better outcomes in many forms of cancer, such as breast and ovarian. As a result, identifying and quantifying the density and location of lymphocytes has gained a lot of interest recently, particularly in the context of identifying which cancer patients to place on immuno-therapy.
Lymphocytes present with a blue tint from the absorption of hemotoxylin, their appearance similar in hue to nuclei, making them difficult to differentiate in some cases. Typically though, lymphocytes tend to be smaller, more chromatically dense, and circular. In this particular use case, our goal was to identify the center of lymphocytes, making this a detection problem (see Section III).
Overview
We break down this approach into 5 steps:
Step 1: Patch Extraction (Matlab): extract patches from all images of both the positive and negative classes
Step 2: Cross-Validation Creation (Matlab): at the patient level, split the patches into a 5-fold training and testing sets
Step 3: Database Creation (Bash): using the patches and training lists created in the previous steps, create 5 sets of leveldb training and testing databases, with mean files, for high performance DL training.
Step 4: Training of DL classifier (Bash): Slightly alter the 2 prototxt files used by Caffe, the solver and the architecture to point to the correct file locations. Use these to train the classifier.
Step 5: Generating Output on Test Images (Python): Use final model to generate the output
There are, of course, other ways of implementing a pipeline like this (e.g., use Matlab to directly create a leveldb, or skip the leveldb entirely, and use the images directly for training) . I’ve found using the above pipeline fits easiest into the tools that are available inside of Caffe and Matlab, and thus requires the less maintenance and reduces complexity for less experienced users. If you have a suggested improvement, I’d love to hear it!
Dataset Description
The dataset consist of 100 images ER+ BCa images scanned at 20x. Each image is 100 x 100. The centers of 3,064 lymphocytes were identified by an expert pathologist.
The format of the files is:
img100_seg.tif: original H&E image
100m.tif: mask of the same size, where a single pixel indicates a lymphocyte center
The data and the associated masks are located here (6.3M).
Examples of these images and their annotates can be seen below






Step 1: Patch Extraction (Matlab)
We refer to step1_make_patches.m, which is fully commented.
At the original 20x magnification, the average size of a lymphocyte is approximately 10 pixels in diameter, much smaller than the 32 x 32 patches used by our network. Since the focus here is on identifying lymphocytes without focusing on the surrounding tissue if the patches were to be extracted at 20x, only 10% of the input pixels would be of interest. The other 90% of the input pixels would eventually learn to be ignored by the network. This would have the unfortunate effect of reducing the discriminative ability of the network. Thus, to increase the predictive power of the system, we artificially resize the images to be 4x as large, so that the entire input space, when centered around a lymphocyte, contains lymphocyte pixels, allowing more of the weights in the network to be useful.
A high level understanding is provided here:
- Positive class exemplars are extracted by randomly sampling locations from a region around the supplied centers of each lymphocyte (generated via the up-scaling).
- The selection of the negative class proceeds as follows (a) a naiıve Bayesian classifier is trained on 1000 randomly selected pixels from the image to generate posterior class membership probabilities for all pixels in the image, (b) for all false positive errors, the distance between the false positive pixels and the closest true positive pixels is computed, (c) iteratively, the pixel with the greatest distance between the false positive and true positive errors is chosen so that negative image patches can be generated from those locations.
 |
 |
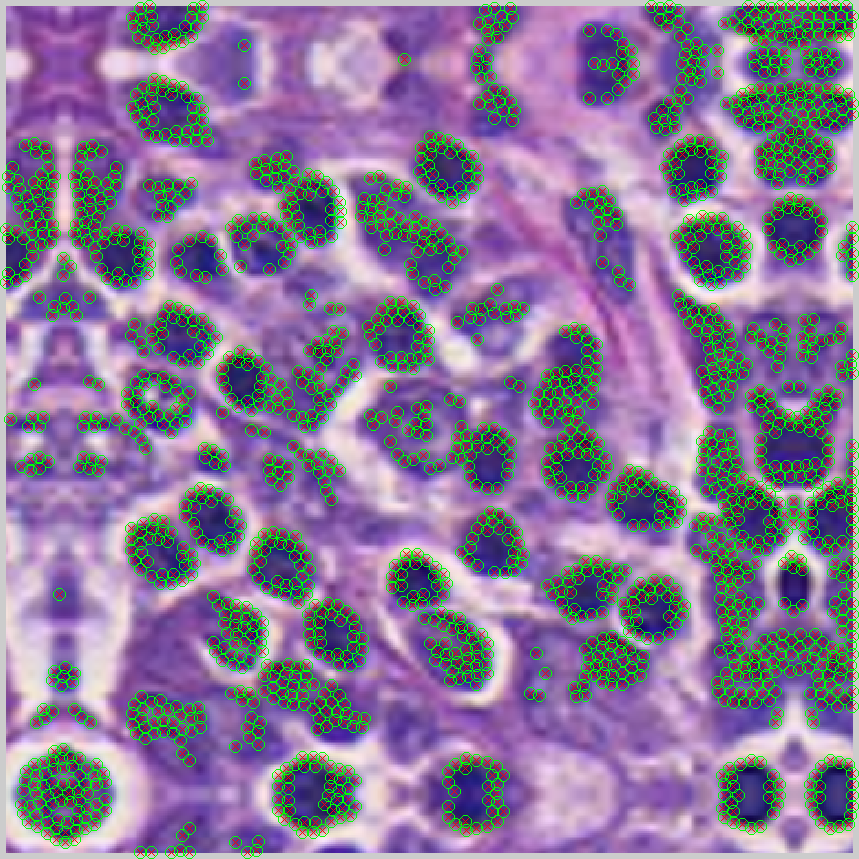 |
| Positive class indicating centers | Naive Bayesian posterior output | Locations selected for negative class |
- From each of these randomly crop patches, save them to disk. At the same time, we maintain a “patient_struct”, which contains all of the file names which have been written to disk.
The file name format for each patch is as follows:
u_v_w_x_y_z.png — > example im1_1_0_n_1_0.png
Where u is the image name, v is the image number (always 1 in this use case), w is the class (0 (not lymphocyte) or 1 (lymphocyte center)), x is the type of patch (“n” is negative, “p” is positive, redundant in this use case with w), y is the number of the patch (1 to 2000), and z is rotation (0, 90, 180, 270 in this case).
Step 2: Cross-Validation Creation (Matlab)
Now that we have all of the patches written to disk, and we have all of the file names saved into patient_struct, we want to split them into a cross fold validation scheme. We use step2_make_training_lists.m for this, which is fully commented.
In this code, we use a 5-fold validation and for each fold, we create 4 text files. Using fold 1 as an example:
train_w32_parent_1.txt: This contains a list of the images which have been used as part of this fold’s training set. This is similar to test_w32_parent_1.txt, which contains the images used for the test set. An example of the file content is:
im1.tif
im17.tif
im33.tif
im91.tif
train_w32_1.txt: contains the filenames of the patches which should go into the training set (and test set when using test_w32_1.txt). The file format is [filename] [tab] [class]. Where class is either 0 (not lymphocyte) or 1 (lymphocyte center). An example of the file content is:
im1_1_p_1_0.png 1
im1_1_p_1_90.png 1
im1_1_p_1_180.png 1
im1_1_p_1_270.png 1
im1_1_p_2_0.png 1
im1_1_p_2_90.png 1
im1_1_p_2_180.png 1
im1_1_p_2_270.png 1
im1_1_p_3_0.png 1
im1_1_p_3_90.png 1
All done with the Matlab component!
Step 3: Database Creation (Bash)
Now that we have both the patches saved to disk, and training and testing lists split into a 5-fold validation cohort, we need to get the data ready for consumption by Caffe. It is possible, at this point, to use an Image layer in Caffe and skip this step, but it comes with 2 caveats, (a) you need to make your own mean-file and ensure it is in the correct format and (b) an image layer can is not designed for high throughput. Also, having 100k+ files in a single directory can bring the system to its knees in many cases (for example, “ls”, “rm”, etc), so it’s a bit more handy to compress them all in to 10 databases (1 training and 1 testing for 5 folds), and use Caffe’s tool to compute the mean-file.
For this purpose, we use this bash file: step3_make_dbs.sh
We run it in the “subs” directory (“./” in these commands), which contains all of the patches. As well, we assume the training lists are in “../”, the directory above it.
Here we’ll briefly discuss the general idea of the commands, while the script has additional functionality (computes everything in parallel for example).
Creating Databases
We use the caffe supplied convert_imageset tool to create the databases using this command:
~/caffe/build/tools/convert_imageset -shuffle -backend leveldb ./ DB_train_1
We first tell it that we want to shuffle the lists, this is very important. Our lists are in patient and class order, making them unsuitable for stochastic gradient descent. Since the database stores files, as supplied, sequentially, we need to permute the lists. Either we can do it manually (e.g., use sort –random) , or we can just let Caffe do it 🙂
We specify that we want to use a leveldb backend instead of a lmdb backend. My experiments have shown that leveldb can actually compress data much better without the consequence of a large amount of computational overhead, so we choose to use it.
Then we supply the directory with the patches, supply the training list, and tell it where to save the database. We do this similarly for the test set.
Creating mean file
To zero the data, we compute mean file, which is the mean value of a pixel as seen through all the patches of the training set. During training/testing time, this mean value is subtracted from the pixel to roughly “zero” the data, improving the efficiency of the DL algorithm.
Since we used a levelDB database to hold our patches, this is a straight forward process:
~/caffe/build/tools/compute_image_mean DB_train_1 DB_train_w32_1.binaryproto -backend leveldb
Supply it the name of the database to use, the mean filename to use as output and specify that we used a leveldb backend. That’s it!
Step 4: Training of DL classifier (Bash)
Setup files
Now that we have the databases, and the associated mean-files, we can use Caffe to train a model.
There are two files which need to be slightly altered, as discussed below:
BASE-alexnet_solver.prototxt: This file describes various learning parameters (iterations, learning method (Adagrad) etc).
On lines 1 and 10 change: “%(kfoldi)d” to be the number of the fold for training (1,2,3,4,5).
On line 2: change “%(numiter)d” to number_test_samples/128. This is to have Caffe iterate through the entire test database. Its easy to figure out how many test samples there are using:
“wc –l test_w32_1.txt”
BASE-alexnet_traing_32w_db.prototxt: This file defines the architecture.
We only need to change lines 8, 12, 24, and 28 to point to the correct fold (again, replace “%(kfoldi)d” with the desired integer). That’s it!
Note, these files assume that the prototxts are stored in a directory called ./model and that the DB files and mean files are stored in the directory above (../). You can of course use absolute file path names when in doubt.
In our case, we had access to a high performance computing cluster, so we used a python script (step4_submit_jobs.py) to submit all 5 folds to be trained at the same time. This script automatically does all of the above work, but you need to provide the working directory on line 11. I use this (BASE-qsub.pbs) PBS script to request resources from our Torque scheduler, which is easily adaptable to other HPC environments.
Initiate training
If you’ve used the HPC script above, things should already be queued for training. Otherwise, you can start the training simply by saying:
~/caffe/build/tools/caffe train –solver=1-alexnet_solver_ada.prototxt
In the directory which has the prototxt files. That’s it! Now wait until it finishes (600,000) iterations. 🙂
Step 5: Generating Output on Test Images (Python)
At this point, you should have a model available, to generate some output images. Don’t worry, if you don’t, you can use mine.
Here is a python script, to generate the test output for the associated k-fold (step5_create_output_images_kfold.py).
It takes 2 command line arguments, base directory and the fold. Make sure to edit line 88 to apply the appropriate scaling; in this case upscale the images to by 4x.
The base directory is expected to contain:
BASE/images: a directory which contains the tif images for output generation
BASE/models: a directory which holds the 5 models (1 for each fold)
BASE/test_w32_parent_X.txt: the list of parent IDs to use in creating the output for fold X=1,2,3,4,5, created in step 2
BASE/DB_train_w32_X.binaryproto: the binary mean file for fold X=1,2,3,4,5, created in step 3
It generates 2 output images for each input. A “_class” image and a “_prob” image. The “_prob” image is a 3 channel image which contains the likelihood that a particular pixel belongs to the class. In this case, the Red channel represents the likelihood that a pixel is a not a lymphocyte center, while the green channel represents the likelihood that a pixel is a lymphocyte center. The two channels sum to 1. The “_class” image is a binary image using the argmax of the “_probs image”.
Notice that the output masks are 400 x 400, or 4x the size of the input masks. This is very important, we trained the classifier at “80x”, so we need to test it at 80x. Since the images are all taken at 20x, we up-sample them by 4x to obtain the correct apparent magnification .
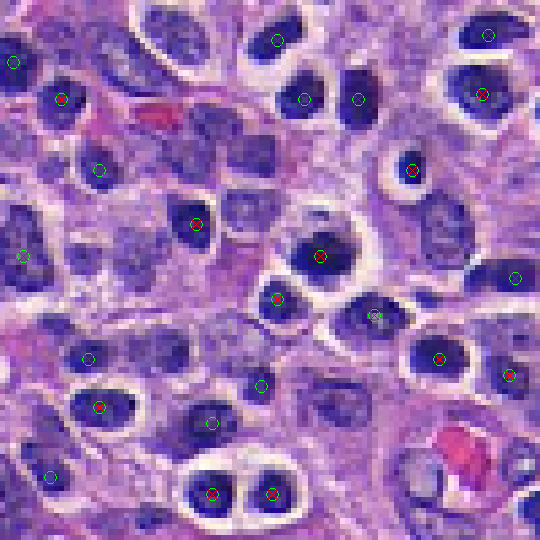 |
 |
| Input Image with Centers Identified | Output with Ground Truth Overlaid |
Typically, you’ll want to use a validation set to determine an optimal threshold as it is often not .5 (which is equivalent to argmax). Subsequently, use this threshold on the the “_prob” image to generate a binary image.
Note that the the posterior probabilities are computed for every pixel in the test image. To identify a singular location most likely to be the center of a lymphocyte, a convolution can be performed using a disk kernel on the _prob image so that the center of the probably regions are highlighted. Iteratively, the highest point in the image is taken as center and a radius is cleared, which is the same size as a typical lymphocyte to prevent multiple centers from being identified for the same lymphocyte. An example of that process is presented here: step5_identify_peak_in_probs.m
Final Notes
Efficiency in Patch Generation
Writing a large number of small, individual files to a harddrive (even SSD) is likely going to take a very long time. Thus for Step 1 & Step 2, I typically employ a ram disk to drastically speed up the processes. Regardless, make sure Matlab does not have the output directory in its search path, otherwise it will likely crash (or come to a halt), while trying to update its internal list of available files.
As well, using a Matlab Pool (matlabpool open), opens numerous workers which also greatly speed up the operation and is recommended as well.
Efficiency in Output Generation
It most likely will take a long time to apply a the classifier pixel wise to an image to generate the output. In actuality, there are many ways to speed up this process. The easiest way in this case is to simply use a color deconvolutional approach to identify regions with low hematoxylin absorption. These regions, which are likely stroma, can be ignored as we know lymphocytes obtain their blue hue from an affinity to hematoxylin.
Keep an eye out for future posts where we delve deeper into this and provide the code which we use!
Magnification
It is very important to use the model on images of the same magnification as the training magnification. This is to say, if your patches are extracted at 80x, then the test images need to be done at 80x as well.
Code is available here
Data is available here (6.3M)
I’m attempting to run step5_create_output_images_kfold.py using the model you provided. When I attempt to run “python step5_create_output_images_kfold.py lymphocyte 3” I receive the following:
https://docs.google.com/document/d/1mY6Nm1DraYaRVgv2Qs0y3acwLYqoMWsJX7qci7e3ZGA/edit?usp=sharing
Have you received this error before? Any thought as the source of the error?
if you notice, they’re not really errors but warnings, so they can be ignored for the near term. to get rid of them entirely, i think you can change the network loading line to include the weights directly as they suggested, with a named parameter of “weights” instead of a positional argument
The third to last line of the output states “This network produces output prob” however, no output is created. I am unsure what the problem is. I have BASE/images, BASE/models, BASE/DB_train_w32_3.binaryproto, and BASE/test_w32_parent_3.txt all downloaded from your github. When ran, the code does create the file to store the output but no output is stored. Do you have an idea as to the problem? Thank you
its likely a misaligned input parameter. i’d recommend stepping through the code step by step, especially when it involves any command line parameters and check that the values you entered are sensible. i say that only because many people have used the provided code without issue :-\
I think your paths are wrong where it get all of the files which start with this patient ID.
How long does running step5_create_output_images_kfold.py on one image usually take? And what is your configuration?
I realize you state above “It most likely will take a long time to apply the classifier pixel wise to an image to generate the output.”
But I am curious how long, numerically, it generally takes on your system. Thank you
i haven’t used this approach in a long time specifically because its too slow (you can can check out this for a faster way which does the same work in <1 minute), but if i remember correctly maybe an hour for a 1k x 1k image? it should be pretty easy to estimate, since each pixel is computed individually, i think an older gpu took 13ms and a newer one 5ms per pixel.
Hello,
In the paper “Deep learning for digital pathology image analysis: A comprehensive tutorial with selected use cases” (http://www.jpathinformatics.org/temp/JPatholInform7129-2991928_081839.pdf) it is mentioned a 40x original magnification on the images:
“At the original ×40 magnification, the average size of a
lymphocyte is approximately 10 pixels in diameter, much
smaller than the 32 × 32 patches used by our network. ”
Here you mention 20x magnification on the images. Which one is the correct?
Thank you !
looking at the images, i would say they are 20x. the original images were scanned at 40x but were downsampled during ROI extraction for this use case to 20x.
by the way, in general, lymphocytes are typically always about the same size. thus it is possible to use them as a ruler to determine approximate magnification. the same cannot be said for e.g., epithelial cells, especially in the context of diseases like cancer.
Hello,
Could you share a little bit more the parameters used to calculate f-score / precision and recall in the paper?
Which distance did you consider from a predicted lymphocyte center (variable test_vals in step5_identify_peak_in_probs.m) and the annotated one (files XXm.tif). Lymphocyte size (radius = 6?)
Thanks!
I likely used a script which looked like this: https://github.com/choosehappy/public/blob/68160ec8ea56d429887d91c94c1d1c1e745149d2/DLLevelTutorial/grade_single_image.m
Hello,
Can you share more information regarding the origin of the dataset used in this model? Did it come from TCGA or other public database? (In the paper I found a mention to breast cancer patients).
Thank you!
Internal dataset that we created for another project, not from the tcga or other public source. i believe it coincides with this work: https://ieeexplore.ieee.org/document/5415659
Hello,
I’m trying to apply this model to TCGA whole images, but it is very expensive (in time) when predicting the 100×100 patches for more than 50k patches in some slides.
Is there a reason to use predict for every pixel on the augmented 432×432 image? (besides the logical one which is related to the fact that the lymphocyte center could be in any pixel in the patch)
Have you tried to reduce (e.g. predicting maybe for every 32 pixels) ?
Thank you!
now-a-days the start of the art is a u-net, which will produce output masks instead of single pixel classifications. there is a post describing that here: